Explaining Conformational Diversity in Protein Families through Molecular Motions - Sergei Gudinin скачать в хорошем качестве
Explaining Conformational Diversity in Protein Families through Molecular Motions - Sergei Gudinin
9 месяцев назад
Не удается загрузить Youtube-плеер. Проверьте блокировку Youtube в вашей сети.
Повторяем попытку...
Повторяем попытку...
Скачать видео с ютуб по ссылке или смотреть без блокировок на сайте: Explaining Conformational Diversity in Protein Families through Molecular Motions - Sergei Gudinin в качестве 4k
У нас вы можете посмотреть бесплатно Explaining Conformational Diversity in Protein Families through Molecular Motions - Sergei Gudinin или скачать в максимальном доступном качестве, видео которое было загружено на ютуб. Для загрузки выберите вариант из формы ниже:
-
Информация по загрузке:
Скачать mp3 с ютуба отдельным файлом. Бесплатный рингтон Explaining Conformational Diversity in Protein Families through Molecular Motions - Sergei Gudinin в формате MP3:
Если кнопки скачивания не
загрузились
НАЖМИТЕ ЗДЕСЬ или обновите страницу
Если возникают проблемы со скачиванием видео, пожалуйста напишите в поддержку по адресу внизу
страницы.
Спасибо за использование сервиса ClipSaver.ru
Explaining Conformational Diversity in Protein Families through Molecular Motions - Sergei Gudinin
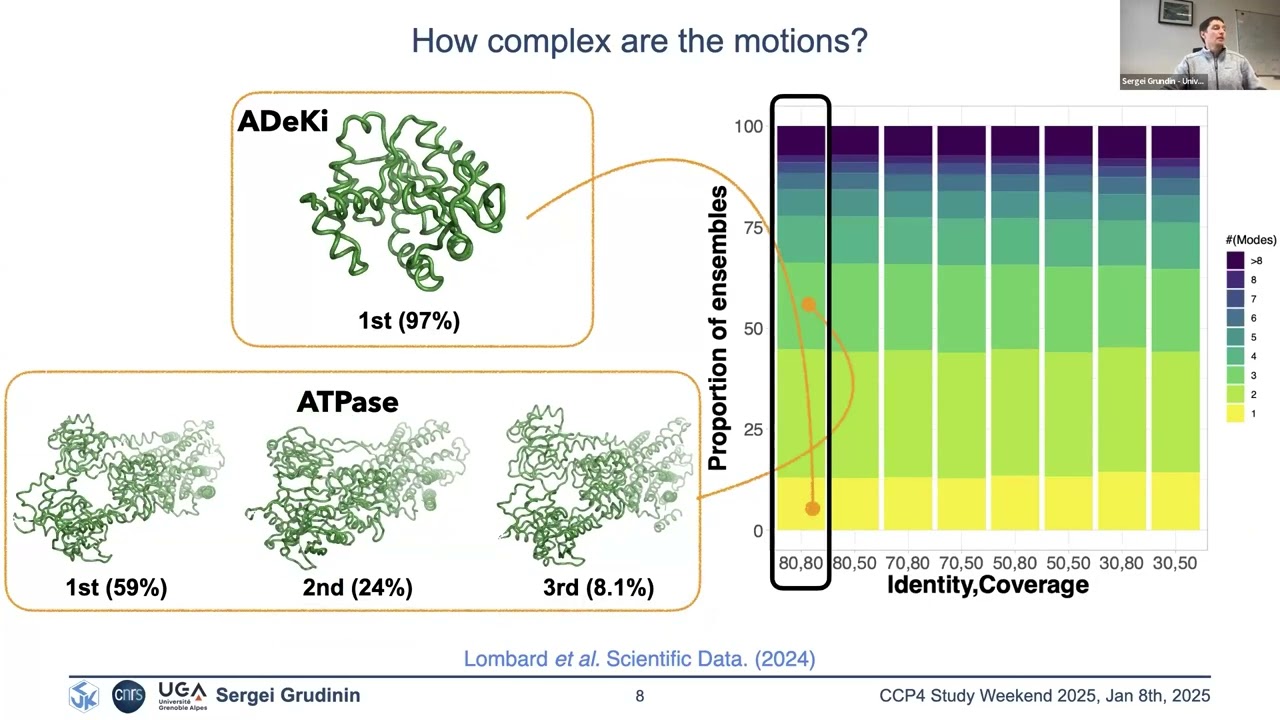
Explaining Conformational Diversity in Protein Families through Molecular Motions and Predicting them with Unsupervised and Supervised Learning Approaches Proteins play a central role in biological processes, and understanding their conformational variability is crucial for unraveling their functional mechanisms. Recent advancements in high-throughput technologies have enhanced our knowledge of protein structures, yet predicting their multiple conformational states and motions remains challenging. I will present our recent work on DANCE and SeaMoon approaches. DANCE is a fully-automated computational pipeline for Dimensionality Analysis for protein Conformational Exploration. DANCE accommodates both experimental and predicted structures. It is suitable for analysing anything from single proteins to superfamilies. Employing it, we clustered all experimentally resolved protein structures available in the Protein Data Bank into conformational collections and characterized them as sets of linear motions (or linear subspaces). The resource facilitates access and exploitation of the multiple states adopted by a protein and its homologs. Beyond descriptive analysis, we applied unsupervised learning and assessed classical dimensionality reduction techniques for sampling unseen states on a representative benchmark. Then, we explored whether continuous compact representations of protein motions could be predicted directly from protein sequences using supervised learning approaches, without exploiting nor sampling protein structures. Our approach, called SeaMoon, leverages protein Language Model (pLM) embeddings as input to a lightweight (∼1M trainable parameters) architecture. SeaMoon achieves a success rate of up to 40% when assessed against ∼ 1 000 collections of experimental conformations exhibiting a wide range of motions. We capture motions not accessible to the normal mode analysis, and generalise to proteins that do not have any detectable sequence similarity to the training set.
Comments