Lab 9: Calculate Band Gap Directly From SCF File Without DOS and Band Structure. скачать в хорошем качестве
Lab 9: Calculate Band Gap Directly From SCF File Without DOS and Band Structure.
5 лет назад
Не удается загрузить Youtube-плеер. Проверьте блокировку Youtube в вашей сети.
Повторяем попытку...
Повторяем попытку...

Скачать видео с ютуб по ссылке или смотреть без блокировок на сайте: Lab 9: Calculate Band Gap Directly From SCF File Without DOS and Band Structure. в качестве 4k
У нас вы можете посмотреть бесплатно Lab 9: Calculate Band Gap Directly From SCF File Without DOS and Band Structure. или скачать в максимальном доступном качестве, видео которое было загружено на ютуб. Для загрузки выберите вариант из формы ниже:
-
Информация по загрузке:
Скачать mp3 с ютуба отдельным файлом. Бесплатный рингтон Lab 9: Calculate Band Gap Directly From SCF File Without DOS and Band Structure. в формате MP3:
Если кнопки скачивания не
загрузились
НАЖМИТЕ ЗДЕСЬ или обновите страницу
Если возникают проблемы со скачиванием видео, пожалуйста напишите в поддержку по адресу внизу
страницы.
Спасибо за использование сервиса ClipSaver.ru
Lab 9: Calculate Band Gap Directly From SCF File Without DOS and Band Structure.
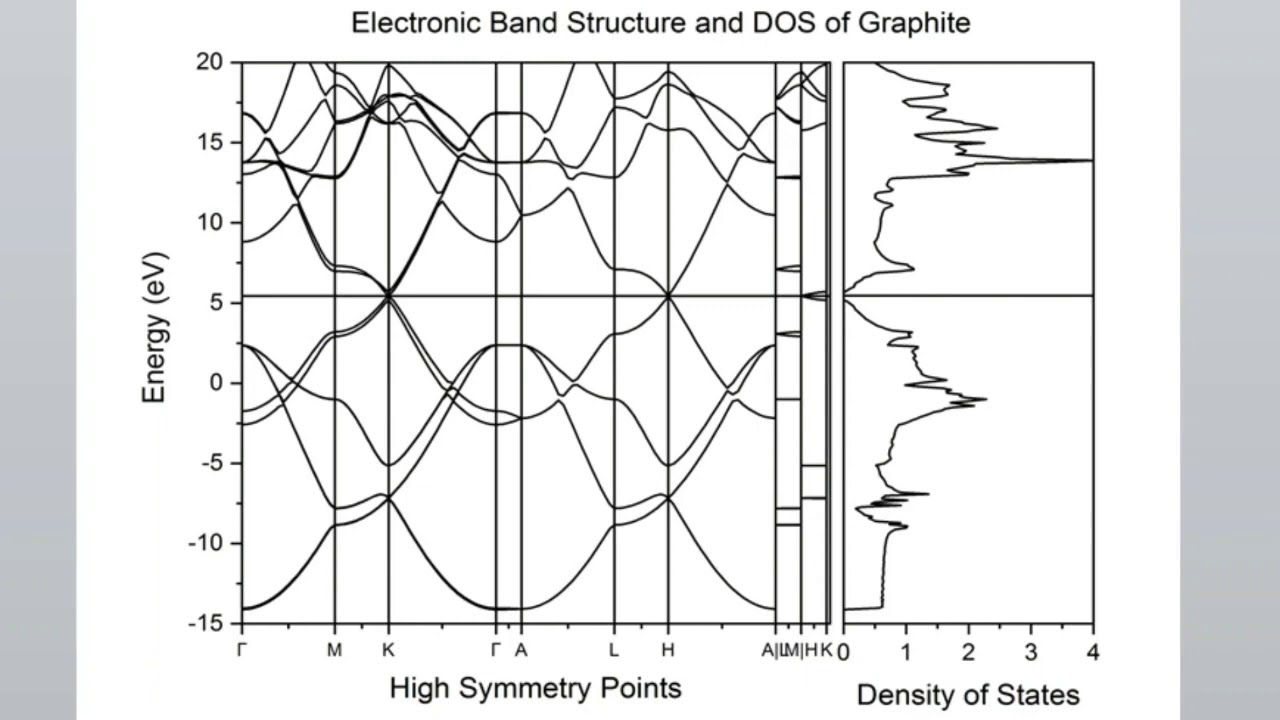
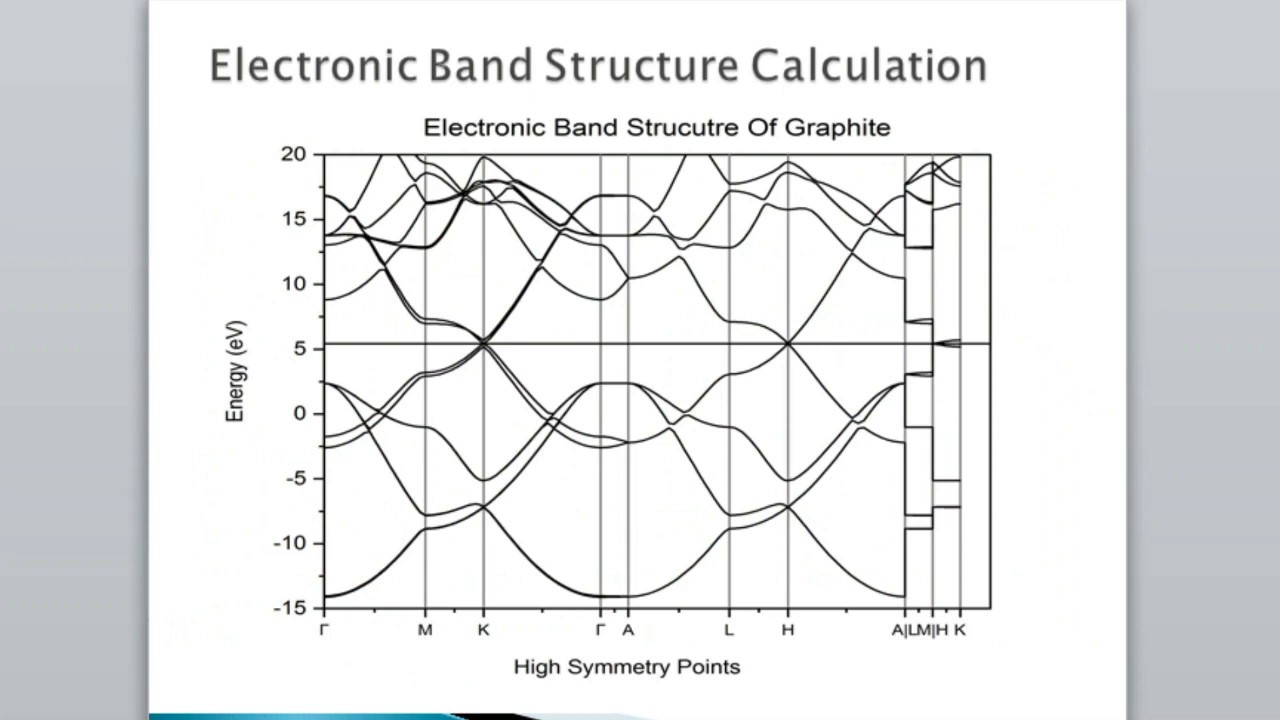
I have tried to make video in a way that ordinary people who are not familiar with Quantum Espresso can understand in common language, I am sorry if there is any error in this video, in this video I have given general information about "Calculate Band Gap Directly From SCF File Without DOS and Band Structure" How to install Quantum-Espresso? : • Lab 1: Installation of Latest Quantum Espr... How to create input file? : (1) • Lab 3.1 : Write input SCF file for Quantum... OR (2) • Lab 3.2 : Generate input SCF file for Quan... How to download pseudo-potential? : • Lab 2: Downloading CIF and Pseudopotential... Download pseudopotentail : https://www.quantum-espresso.org/upf_... https://www.quantum-espresso.org/upf_... Diamond SCF : diamond.in ====================== &CONTROL calculation = 'scf' pseudo_dir = '.' disk_io = 'none' / &SYSTEM a = 3.57371 degauss = 0.01 ecutrho = 400 ecutwfc = 40 ibrav = 2 nat = 2 ntyp = 1 nbnd = 6 ! 2 X 4 valence electrons = 8 valence electrons = 4 kohn sham states + 2 occupations = "fixed" smearing = "gaussian" / &ELECTRONS / ATOMIC_SPECIES C 12.0107 C.pbe-n-rrkjus_psl.1.0.0.UPF ATOMIC_POSITIONS crystal C 0.7500000000 0.7500000000 0.7500000000 C 0.5000000000 0.5000000000 0.5000000000 K_POINTS automatic 6 6 6 1 1 1 Si SCF : si.in ========== &CONTROL calculation = "scf" pseudo_dir = "." disk_io = 'none' / &SYSTEM a = 5.46873 degauss = 0.01 ecutrho = 400 ecutwfc = 40 ibrav = 1 nat = 8 ntyp = 1 nbnd = 18 ! 8 X 4 valence electrons = 32 valence electrons = 16 kohn sham states + 2 occupations = "fixed" smearing = "gaussian" / &ELECTRONS / K_POINTS {automatic} 4 4 4 1 1 1 ATOMIC_SPECIES Si 28.08550 Si.pbe-n-rrkjus_psl.1.0.0.UPF ATOMIC_POSITIONS {crystal} Si 0.000000 0.000000 0.000000 Si 0.750000 0.750000 0.250000 Si 0.000000 0.500000 0.500000 Si 0.750000 0.250000 0.750000 Si 0.500000 0.000000 0.500000 Si 0.250000 0.750000 0.750000 Si 0.500000 0.500000 0.000000 Si 0.250000 0.250000 0.250000
Comments


![Расчеты SCF с использованием BURAI (графический интерфейс для Quantum ESPRESSO) - [УРОКИ №4]](https://imager.clipsaver.ru/CZS2In5PQqg/max.jpg)






![Calculate the BAND-GAP from SCF calculation - QUANTUM ESPRESSO [Tutorial]](https://imager.clipsaver.ru/tsgcceURNG8/max.jpg)