Solvation of a protein in a water box by using VMD скачать в хорошем качестве
Solvation of a protein in a water box by using VMD
6 лет назад
Не удается загрузить Youtube-плеер. Проверьте блокировку Youtube в вашей сети.
Повторяем попытку...
Повторяем попытку...

Скачать видео с ютуб по ссылке или смотреть без блокировок на сайте: Solvation of a protein in a water box by using VMD в качестве 4k
У нас вы можете посмотреть бесплатно Solvation of a protein in a water box by using VMD или скачать в максимальном доступном качестве, видео которое было загружено на ютуб. Для загрузки выберите вариант из формы ниже:
-
Информация по загрузке:
Скачать mp3 с ютуба отдельным файлом. Бесплатный рингтон Solvation of a protein in a water box by using VMD в формате MP3:
Если кнопки скачивания не
загрузились
НАЖМИТЕ ЗДЕСЬ или обновите страницу
Если возникают проблемы со скачиванием видео, пожалуйста напишите в поддержку по адресу внизу
страницы.
Спасибо за использование сервиса ClipSaver.ru
Solvation of a protein in a water box by using VMD
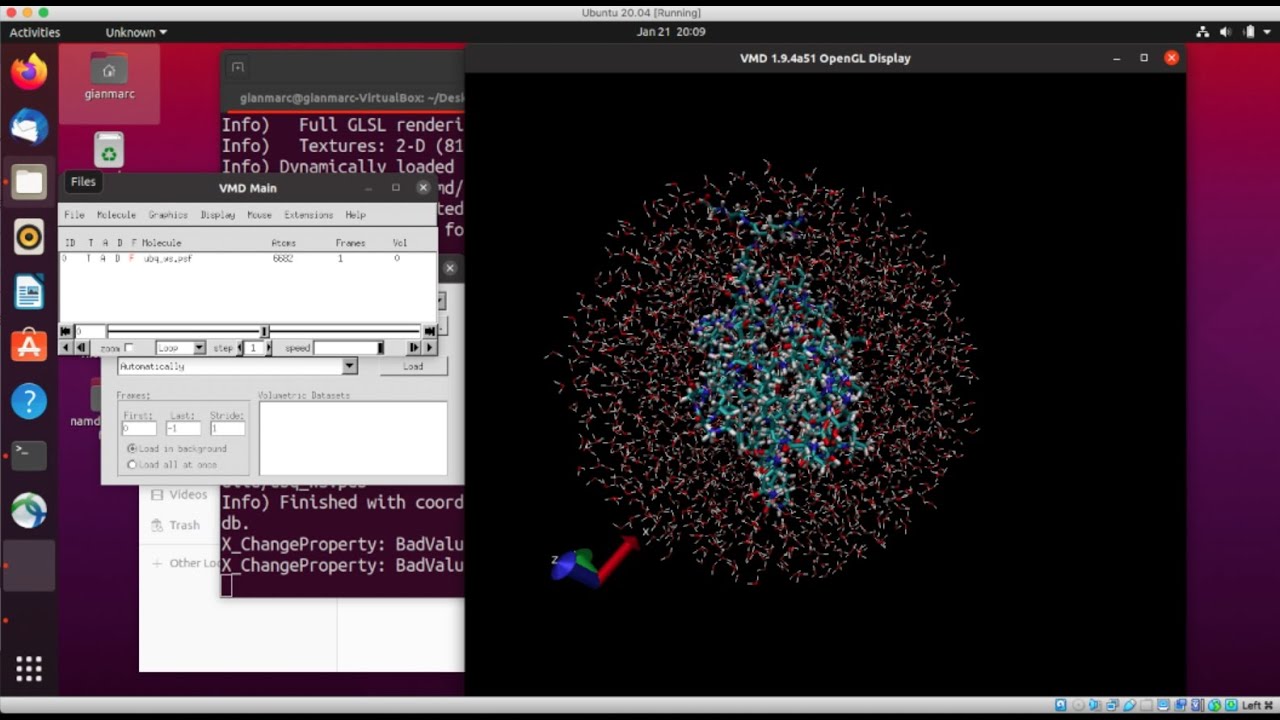
This video explains how to use VMD to solvate a protein in a water box which is an essential step for molecular dynamic simulations. The Solvate plugin provides both a graphical user interface and text commands for automatic solvation in VMD. Using psfgen and VMD commands, the plugin performs the following functions: Generate water block coordinates (VMD) Replicate water block (psfgen) Combine water block and solute (psfgen) Create a PDB with just the water you want (VMD) Merge the solute and the cutout water (psfgen) Graphical User Interface The Solvate interface is fairly straightforward. Simply fill in the values and click the "Solvate" button to perform the solvation. Files are created in the current working directory, as in the command-line interface. PSF and PDB filenames are not required if "Waterbox Only" is selected, and the box size is ignored and the minmax of the molecule is used if "Use Molecule Dimensions" is selected. All other fields are required, although default values are automatically filled-in when the plugin is launched. Note that "Use Molecule Dimensions" cannot be selected if "Waterbox Only" is. The "Rotate to minimize volume" check button toggles whether or not the system should be rotated to minimize the amount of water needed to fulfill the given boundary conditions prior to solvation. Because the solvation box must be oriented along the cardinal axes, this can result in significant system size reductions, but should not be used if the initial orientation was chosen to facilitate analysis. The parameters "Rotation Increment" and "Selection for Rotation" control the number of degrees between attempted rotations and the portion of the molecule used for calculating these rotations, respectively.
Comments